
阿尔茨海默病(Alzheimer'sdisease,AD)是一种以认知功能障碍为主要症状的神经系统退行性疾病。随着老龄化社会的到来,AD的发病人数逐年增加,预计到2050年全球确诊的AD患者将达到1.52亿人,已成为21世纪威胁人类健康的最严重疾病之一[1]。有研究表明,AD早期神经网络兴奋性异常能够导致认知功能障碍[2]。电压门控性钠离子通道(voltage-gatedsodiumchannel,简称VGSC或Nav)参与可兴奋细胞动作电位形成,直接影响神经网络兴奋性。作为中枢兴奋性神经元钠通道的主要亚型Nav1.6,主要特征为持续电流和复苏电流而致的重复性神经元放电。持续钠电流可增加阈下刺激所致神经元异常放电的可能性,复苏钠电流使神经元很易产生快速自发兴奋[3,4]。研究团队前期发文表明抗癫药拉莫三嗪应用可改善AD认知障碍[5],于是作者推测Nav1.6有可能介导神经网络超兴奋,在AD认知障碍中发挥重要作用。
2022年3月30日,大连医科大学基础医学院的李韶团队与苏州大学神经科学研究所的马全红团队合作在生物学TOP期刊AgingCell发表了题为“ReducingNav1.6expressionattenuatesthepathogenesisofAlzheimer'sDiseasebysuppressingBACE1transcription”的研究论文。首次发现APP/PS1小鼠(AD模型鼠)脑内Nav1.6水平随着年龄的增长明显增高,下调海马Nav1.6表达可通过影响BACE1的转录减少Aβ的生成,改善AD的病理过程。

Nav1.6由SCN8A基因编码,在兴奋性神经元轴突始段的郎飞氏结聚集,此部位是动作电位产生处[6]。Nav1.6通过调节持续电流、再生电流和重复神经元放电在神经兴奋性中发挥重要作用。研究团队之前的研究发现,淀粉样肽Aβ的代谢前体蛋白(APP)与Nav1.6在郎飞氏结和轴突始段共定位[7];APP通过Go-coupledJNK通路增强Nav1.6电流[8];Nav1.6的钠电流密度因Aβ1-42低聚物的存在而增强[9]。也有研究表明,Nav1.6是Aβ1-42诱导的癫痫样放电和海马过度兴奋的关键参与者[10]。所有这些研究都提示Nav1.6参与了AD的发病过程。但是Nav1.6在AD发病机制中的具体作用还未见报道。
研究者首先检测了不同月龄的野生型(WT)和APP/PS1转基因小鼠海马内总蛋白及膜蛋白Nav1.6和Nav1.2表达水平。结果发现2-3月龄的APP/PS1小鼠和WT小鼠相比,脑内总蛋白及膜蛋白Nav1.6以及Nav1.2的蛋白水平都无明显差异。而7-8月龄的APP/PS1小鼠脑内总蛋白及膜蛋白的Nav1.6蛋白水平都明显高于同月龄的WT小鼠且膜蛋白Na1.6增加的更为明显,但Nav1.2的蛋白水平却无明显变化(图1),表明Nav1.6在AD的病理发展中发挥重要作用。
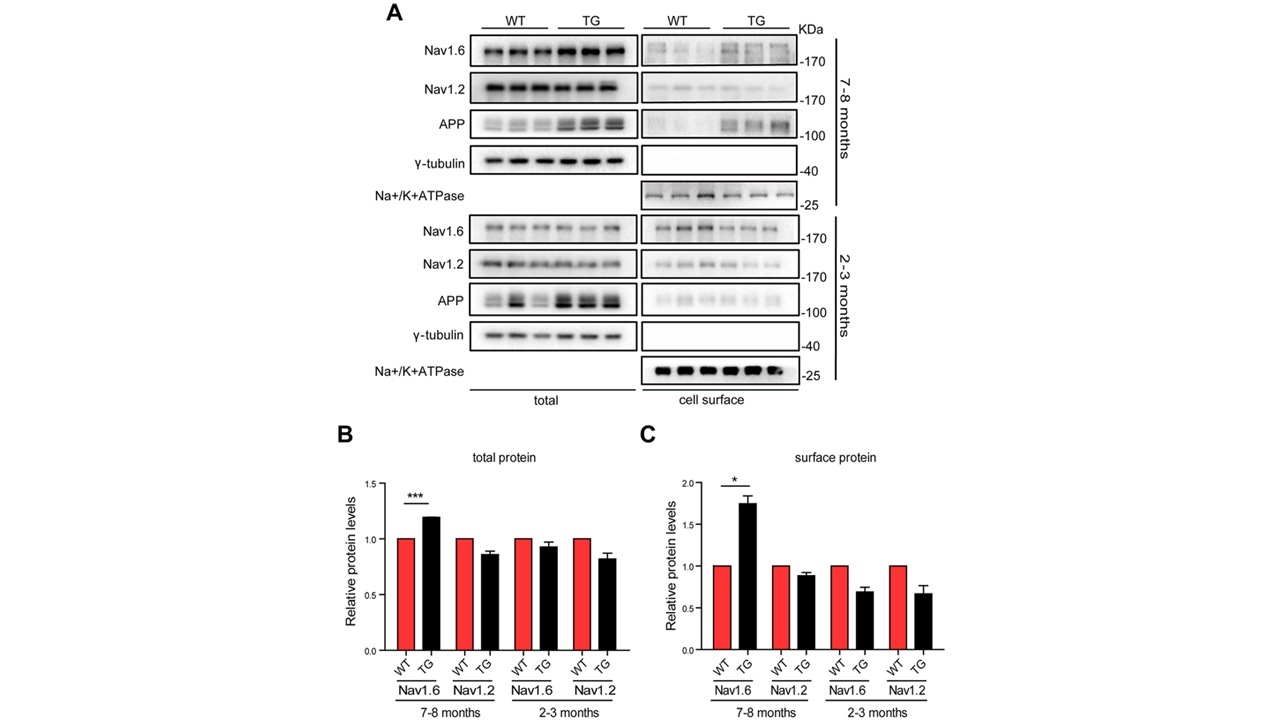
图1Nav1.6在不同月龄小鼠海马内的表达(图源:De-JuanYuanetal.,AgingCell,2022)
由于Nav1.6在老年APP/PS1小鼠大脑中的表达显著增加,研究者推测敲低Nav1.6表达有可能缓解AD样症状。因此他们将编码Nav1.6的shRNA腺相关病毒注射到5月龄的APP/PS1转基因小鼠的双侧海马体中,敲减其海马内Nav1.6表达。病毒注射三个月后利用行为学、高尔基染色、电生理学等实验方法,阐明了敲低APP/PS1小鼠海马内Nav1.6表达能够改善其认知缺陷,减轻突触损伤,降低神经元的兴奋性(图2)。
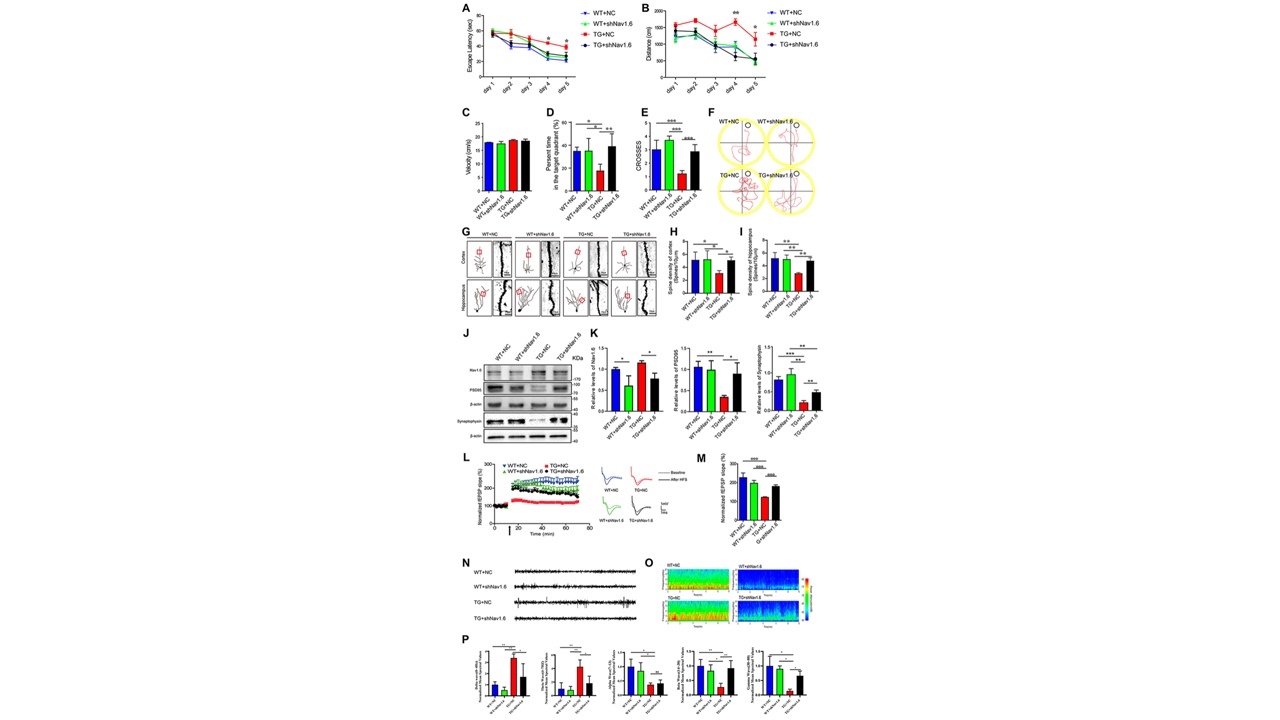
图2在APP/PS1小鼠脑内敲低Nav1.6表达改善认知缺陷、减轻突触损伤以及降低神经元兴奋性(图源:De-JuanYuanetal.,AgingCell,2022)
接下来研究者探索了敲减Nav1.6对突触可塑性和认知功能发挥有益作用的具体机制。有研究表明,神经元过度兴奋与Aβ斑块的积累有关[11],但其机制仍然未知。在此,研究者通过免疫组化染色、蛋白印迹、细胞培养等方法,发现敲低Nav1.6之后Aβ斑块减少,APP剪切酶BACE1及代谢产物β-CTF表达下降(图3)。这表明敲低Nav1.6通过抑制BACE1转录从而减少β分泌酶对APP的剪切,进而减少Aβ的产生。
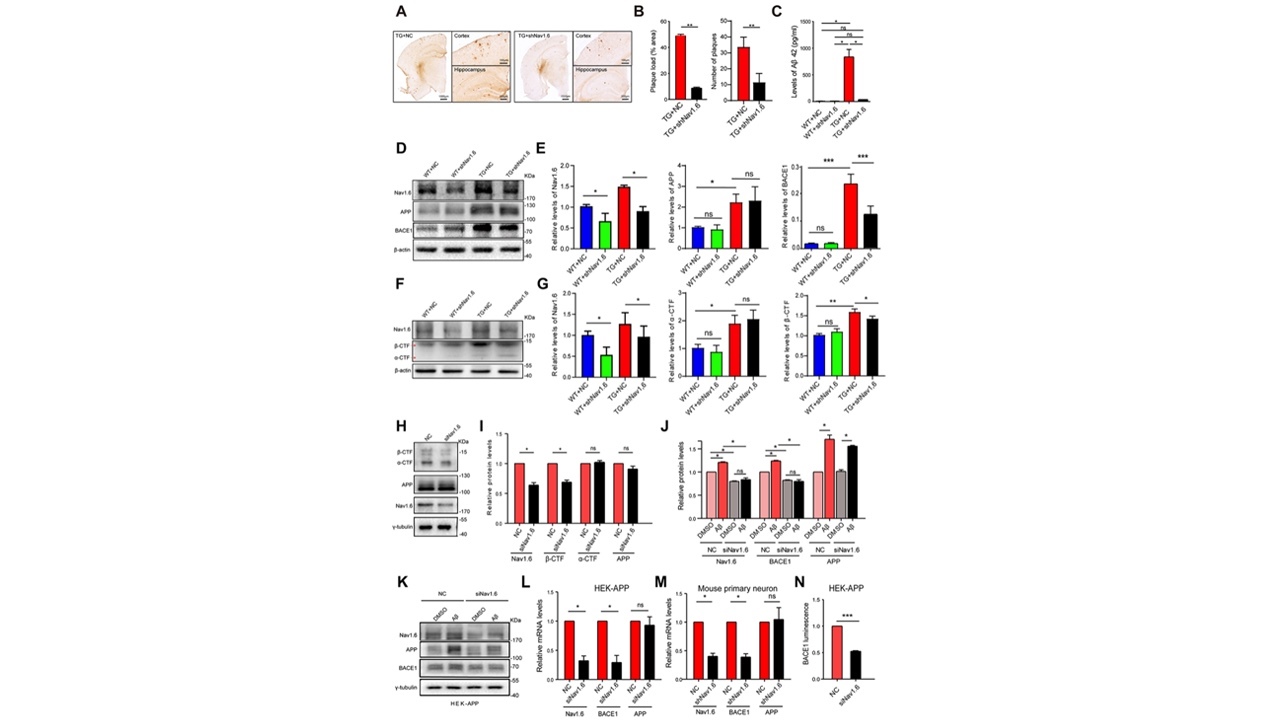
图3敲低Nav1.6影响BACE1表达、减少Aβ产生(图源:De-JuanYuanetal.,AgingCell,2022)
Nav1.6作为钠通道的一个亚型,通过Na+内流参与各种生物过程。Nav1.6调节BACE1转录是否依赖于它作为离子通道的功能特性。为了探究这种可能性,研究者培养了HEK-Nav1.6及HEK-APP细胞系,给予Aβ刺激,利用TTX特异性阻断钠通道,利用siNav1.6敲减Nav1.6表达。发现Nav1.6对BACE1转录的调节取决于其通道特性,并且该调节依赖于Aβ存在的前提下(图4)。
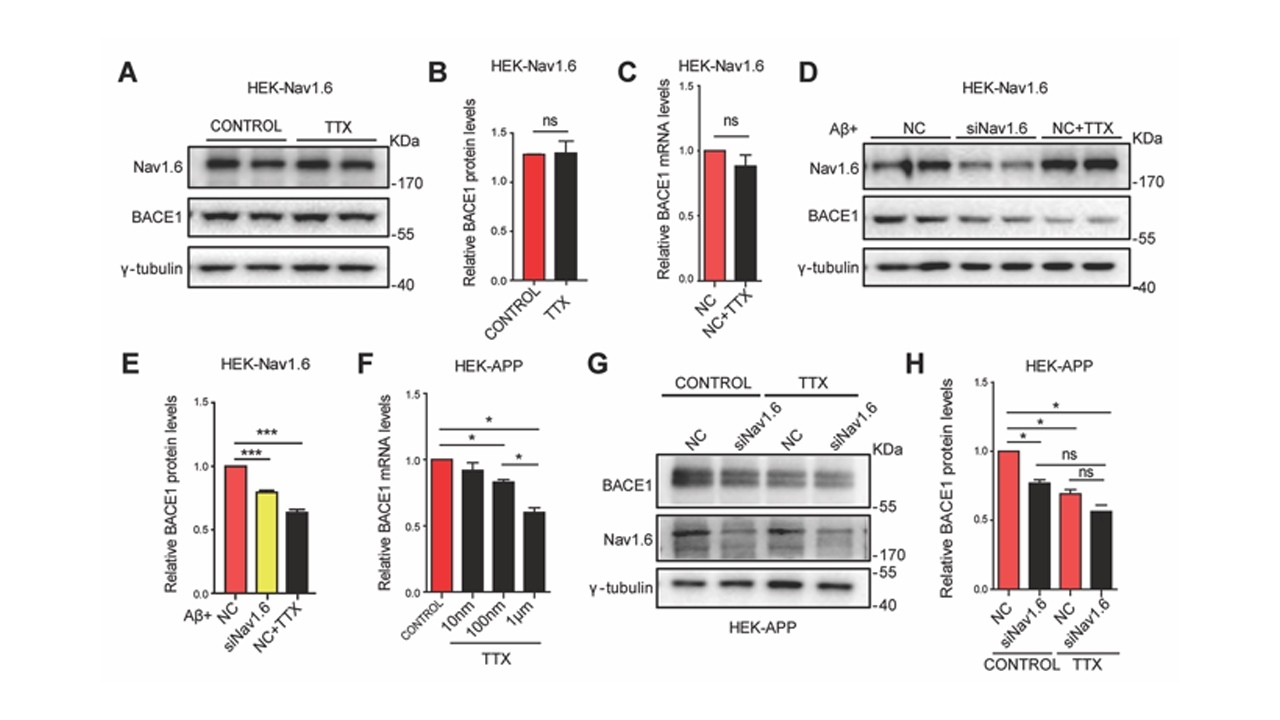
图4Nav1.6对BACE1转录的调节取决于其通道特性(图源:De-JuanYuanetal.,AgingCell,2022)
细胞内Ca2+升高是AD的早期病理改变并加速Aβ的产生[12],当大量的Na+内流时,会使钠钙交换体发生反向转运,从而使大量钙离子内流,导致钙离子超载,轴突损伤,而引起这种病理状态发生的持续内流的Na+部分来源于Nav1.6通道[13]。活化的T细胞核内因子(NFAT)为细胞信号转导中重要的转录因子,NFAT的活化主要受细胞内钙/钙调神经磷酸酶刺激,启动脱磷酸过程进行核移位,调节BACE1的转录[14]。研究者进一步发现敲低Nav1.6能够抑制钠钙反向交换减轻细胞内钙超载从而增加非活化NFAT1水平减少BACE1转录(图5)。
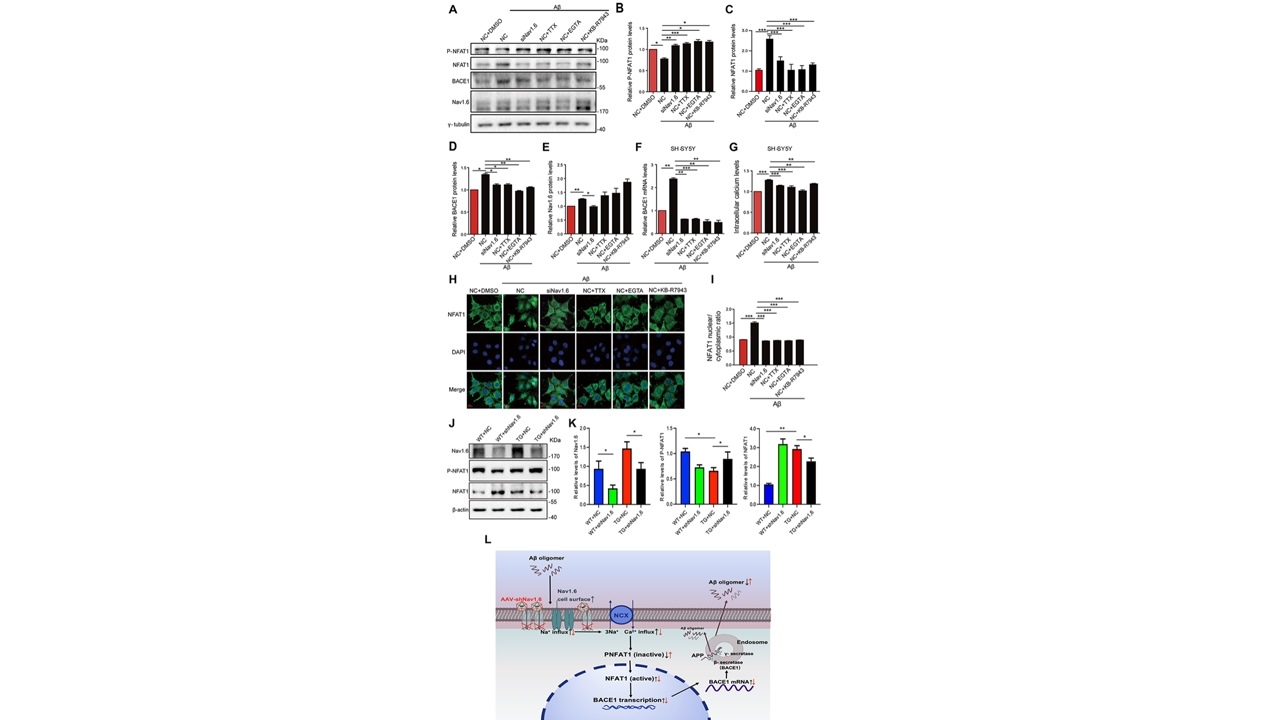
图5Nav1.6对BACE1转录的调节取决于其通道特性(图源:De-JuanYuanetal.,AgingCell,2022)
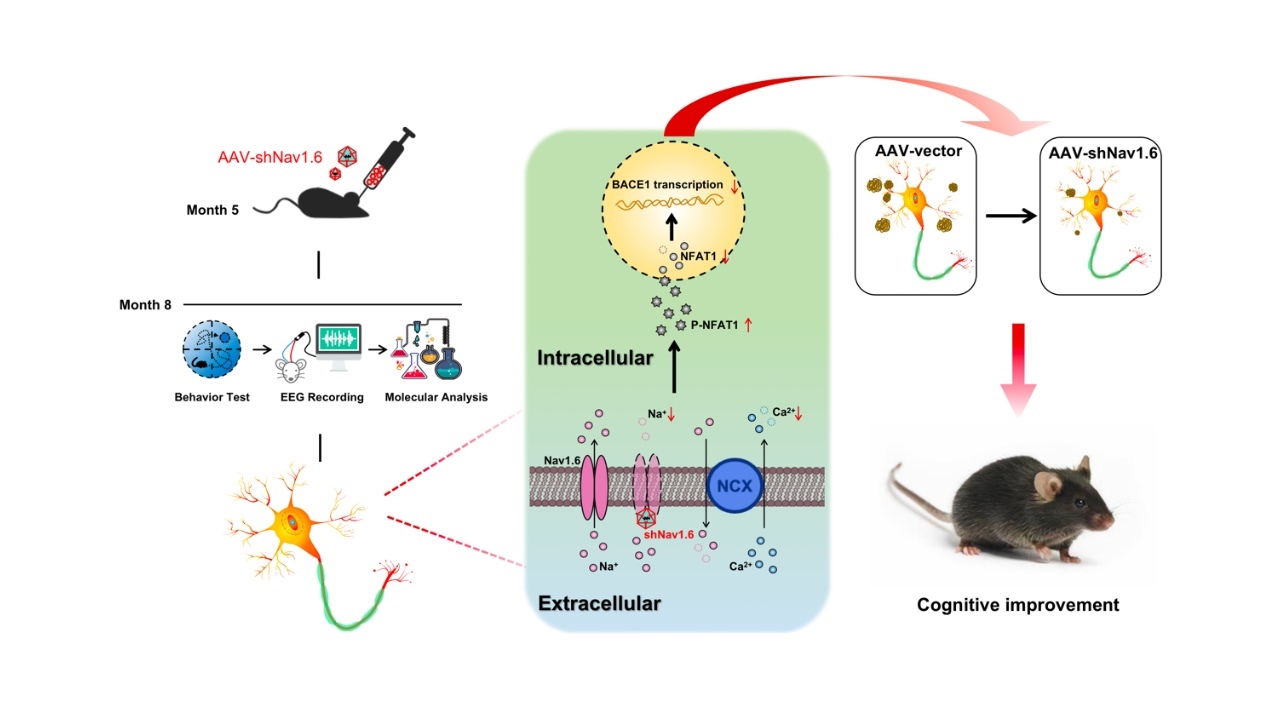
图6工作总结图:下调Nav1.6表达通过影响BACE1的转录减少Aβ的生成(图源:De-JuanYuanetal.,AgingCell,2022)
综上所述,研究者将APP/PS1小鼠作为AD模型鼠,利用AAV病毒定点敲低其海马Nav1.6表达。通过行为学、形态学及电生理学研究证明,敲减Nav1.6可以减低BACE1的转录水平,这一过程依赖于Aβ寡聚体诱发的毒性作用(图6)。其具体的机制为下调Nav1.6表达能够抑制钠钙交换体反向转运,减少细胞内钙超载,从而增加非活性NFAT1水平,减少BACE1的转录,导致APP/PS1转基因小鼠的Aβ水平降低,突触损伤减轻,改善APP/PS1小鼠的学习和记忆障碍(图6)。然而这项研究还存在一些有待解决的问题,目前世界上还没有靶向Nav1.6的药物,因此有待开发针对Nav1.6离子通道的特异阻断剂。该研究首次阐明下调Nav1.6表达改善AD的病理过程,提示Nav1.6可作为治疗AD的潜在靶点,具有较高的创新性,为后续AD的诊断和药物研发提供新的路径。
原文链接:https://onlinelibrary.wiley.com/doi/10.1111/acel.13593
来源:逻辑神经科学
作者:武薇,李韶

